|
This
is a contribution from members of THINCS,
Your number is up! How
cholesterol, homocysteine, and infections conspire to cause heart disease
and
stroke Kilmer
S. McCully M.D. Your essayist spent 40 years, the major part of his professional career,
pursuing the connection between homocysteine, cancer, arteriosclerosis, and
diseases of aging. The purpose
of this essay is to discuss the way in which cholesterol, homocysteine, and
infections interact in the cells and tissues of the body to promote these
serious diseases. This
essay will focus on how these factors conspire to create the vulnerable
plaques of atherosclerosis in the lining of the arteries. When vulnerable
plaques rupture, the results are hemorrhage into the plaque and thrombosis (blood
clot) causing occlusion of the lumen of the artery. This disastrous
complication is the direct cause of heart attack, stroke, and amputation,
depending upon which artery is affected. Cholesterol Cholesterol [chole = bile, and stereos = solid, Greek] is a fatlike
alcohol occurring in animal fats and oils, especially gallstones, bile,
blood, brain, milk, egg yolk, liver, kidney, nerves, atherosclerotic
arteries, and adrenal glands. Cholesterol
is a lipid, a substance extractable from tissues by organic solvents like
chloroform-methanol. When the
lipid fraction is treated with alkali, triglycerides, phospholipids,
sphingolipids and lipoproteins are converted to substances soluble in water.
Because of its chemical composition, however, cholesterol remains
with the lipid fraction after alkali treatment, hence it is a
non-saponifiable lipid. Cholesterol
is a fatty alcohol that is a normal constituent of all animal cell membranes
and is not present in plant tissues. In
the liver, intestine and other tissues cholesterol is synthesized from
acetate in the coenzyme A form, through a complex series of reactions
involving several terpene intermediates, as demonstrated by Konrad Bloch and
other investigators. In the mid 19th century the important German pathologist
Rudolf Virchow described the process by which arteriosclerosis develops in
arteries. His name for the disease was “endoarteritis chronica
deformans nodosa.” He
described inflammatory changes in arterial wall, fatty infiltration of
intima, mucoid degeneration of arterial wall, fibrosis, calcification, and
atheroma with crystal deposition. He
suggested that altered permeability of arterial intima led to increased
filtration of plasma and deposition of plasma fats in association with the
degenerative changes of arterial wall.
In 1914 Ludwig Aschoff described deposition of cholesterol crystals
in aortic atheromas. Wladimir Sergius Ignatowsky, an obscure medical investigator in St.
Petersburg, became interested in the idea that arteriosclerosis in
populations in England was related to consumption of meat, eggs and dairy
products by the wealthier classes. In
1908 he reported the results of feeding these foods to rabbits, a vegetarian
species. He discovered plaques
in the arteries of rabbits resembling the plaques found in human arteries
with atherosclerosis, including deposition of lipids and cholesterol
crystals. Because of the large
amounts of animal protein in the experimental diet, he suggested that the
plaques were caused by protein intoxication.
In 1914 another investigator in St. Petersburg, Nicolai Anitschkow,
decided to feed purified cholesterol dissolved in plant oils to rabbits, and
he discovered deposition of cholesterol in many organs including the
arteries of these animals. He
suggested that the plaques in Ignatowsky’s rabbits were caused by the
cholesterol of the animal foods in the experimental diet.
These experiments were undoubtedly related to the discovery of
cholesterol crystals in human plaques by Aschoff and other investigators. In America a young investigator in Boston named Harry Newburgh, who was
trained in medical research at Massachusetts General Hospital, was recruited
by the University of Michigan Medical School in 1919 to become the first
full time faculty member in America devoted to research in clinical
medicine. He decided to repeat Ignatowsky’s experiments with rabbits
using meat powder as a source of protein in the diet from which all fats and
cholesterol had been removed by extraction with organic solvents.
He found that plaques developed in the arteries of his rabbits, even
though no fats or cholesterol were present in the experimental diet,
effectively disproving Anitschkow’s suggestion that these substances
caused the arterial plaques. Newburgh
then started giving individual amino acids one by one to dogs, rabbits and
other animals, looking for evidence of arterial plaques.
Although he found no plaques, he discovered toxicity of several amino
acids to kidneys, leading to the Newburgh-Marsh diet, an effective treatment
for kidney failure in the years before dialysis treatment.
The amino acids methionine and homocysteine had not been discovered
when Newburg conducted his experiments, and if they had been available, he
would have discovered the ability of these amino acids to cause plaques in
animals. According to Ancel Keys, a professor of physiology and nutrition in
Minnesota, the earliest epidemiological investigation relating cholesterol
to human atherosclerosis was conducted by the Dutch physician, CD DeLangen,
who reported his experience working with patients in Java.
In 1916 DeLangen reported that his patients had low levels of
cholesterol in the blood, possibly explaining the rarity of atherosclerosis,
phlebothrombosis and gallstones in this Asian population.
In contrast, Javanese stewards on Dutch ships in Java, who consumed
the rich Dutch diet, had increased levels of cholesterol in their blood, and
DeLangen advocated a low cholesterol diet for prevention of atherosclerosis,
based on these observations. Keys later became the leading advocate of what became to be known as the
“diet-heart” hypothesis that related dietary consumption of fats and
cholesterol to susceptibility to vascular disease, especially coronary heart
disease. Keys and his colleagues studied mortality rates from
countries around the world, reporting an association with dietary fat
available for consumption in the “Seven Countries Study.”
However, when similar data from other sources, available to Keys, are
considered, the association becomes quite weak. For example, the mortality rate in Finland was almost 7 times
higher than in Mexico, although the fat consumption was identical.
Subsequently authoritative critical review of the Seven Countries
Study by statisticians Smith and Pinckney revealed “a massive set of
inconsistencies and contradictions,” leading to the conclusion that the
“study cannot be taken seriously by the objective and critical
scientist.” The longest running study of cardiovascular disease in a population was
initiated in Framingham, Massachusetts in 1948 and continues to this day. This landmark longitudinal study identified important major
risks for disease, especially smoking, lack of exercise, age, male gender,
and elevated cholesterol levels in younger men.
In spite of the great emphasis on cholesterol levels, the Framingham
study made several critical observations that refute the “diet-heart”
hypothesis. In the first place,
dietary cholesterol has no relation to cholesterol levels in the blood, and
dietary cholesterol has no relation to the risk of developing cardiovascular
disease. This observation was
confirmed by multiple large studies from Chicago, Puerto Rico, Honolulu,
Netherlands, Ireland, and the massive Lipid Research Clinics study of US
citizens. The next astounding
finding is that elevated cholesterol is not a risk factor for women of any
age or for men over age 47. Furthermore,
both total mortality and cardiovascular mortality in Framingham participants
increases in those with LOW cholesterol levels.
This finding has been confirmed by multiple studies from Canada,
Sweden, Russia, and New Zealand. These
contradictory findings have been ignored, distorted, and incorrectly
reported by supporters of the “diet-heart” hypothesis. The massive Multiple Risk Factor Intervention Trial (MRFIT) screened
360,000 men to find those with the highest risk of developing cardiovascular
disease. Approximately 12,000 overweight, hypertensive, smokers with
elevated cholesterol levels were recruited for this 7 year trial, involving
consuming a low fat diet, smoking cessation, exercise and anti-hypertensive
drugs. At the end of the trial,
blood pressure was down, smoking decreased, and average cholesterol levels
were down 7%. When the results
of this $100M trial were analyzed, 115 in the treatment group had died of
heart disease, compared with 124 in the control group, an insignificant
difference. Looking at
mortality from all causes, there were 265 deaths in the treatment group,
compared with 260 in the control group. In
looking at the failure of this massive and expensive $100M trial, the
investigators found minor benefits of smoking cessation, no benefit of
lowering blood pressure, and no effect of lowering cholesterol levels by 2%
compared with the control group. In the even more massive Lipid Research Clinics (LRC) trial, 4000
participants with very high cholesterol levels were selected from almost
half a million men. After
significant lowering of cholesterol levels for 7 years by the resin
cholestyramine, 190 men had suffered nonfatal heart attacks in the treatment
group, compared with 212 in the control group.
As for fatal heart attacks, the figures were 1.7% compared with 2.3%,
a difference of 0.6%, or 12 individuals.
The investigators expressed these differences as relative risk
reductions of 19% and 30% by throwing out the denominators of their
fractions. In the later trials with statin drugs that lower cholesterol levels more
effectively than the unpleasant resin cholestyramine, a similar statistical
approach was taken to increasing the apparent effect on reducing
cardiovascular mortality and adverse events.
In an analysis of 6 major statin trials (EXCEL, 4S, WOSCOPS, CARE,
AFCAPS, LIPID), the reduction of cardiovascular mortality ranged from -19%
to -41% when expressed as relative risk reduction, but from –0.12% to
-3.5% when expressed as absolute risk reduction.
This statistical manipulation to make the results more impressive
illustrates Mark Twain’s aphorism: “There
are lies, damn lies, and statistics.”
Thus a multi-billion dollar drug industry depends upon using
misleading interpretations of statistics showing trivial differences between
treated and control groups. Another way of looking at absolute risk reduction is to consider the
number needed to treat (NNT) to achieve one positive outcome.
In the case of antibiotic therapy for pneumonia the NNT is 1.1,
meaning that 10 of 11 patients treated are cured, or a 90% success rate. The NNT for a successful medication is generally considered
to be 2 to 4, meaning a 25% to 50% success rate.
In the recent ENHANCE trial of Vitorin, a combination of simvistatin
and ezetimibe prescribed to lower blood cholesterol, the NNT is 67, meaning
that a positive outcome is observed in only one of 67 participants when
taken for 5 years. This number
corresponds to a 2% success rate, or a 98% failure rate, a dismal and
unacceptable outcome. In
addition, Vitorin failed to prevent intimal-medial thickening, as assessed
by ultrasound, contradicting the claim that cholesterol lowering by this
medication prevents the early stages of arteriosclerosis.
These results further call into question the claims for a preventive
effect of cholesterol lowering on arteriosclerosis. The gigantic MONICA study, sponsored by the World Health Organization,
analyzed the relation between cardiovascular mortality and blood cholesterol
in 27 countries, in much the same way as the Seven Countries Study.
The results are similar, showing that countries like Japan and China
have low mortality and low cholesterol levels, and countries like Finland
have high mortality and high cholesterol levels.
Yet countries like France, Germany, Switzerland, and Luxembourg have
a low mortality rate and yet a high blood cholesterol value.
This so-called “French paradox” is not a paradox at all, when
examination of the data reveals great disparities in mortality between
different regions with the same cholesterol levels.
Similarly the residents of Corfu have a 5 fold greater mortality than
residents of Crete, despite identical dietary practices and identical
cholesterol levels. Residents
of the North Karelia regions of Finland have mortality rate of 493/100,000
and those in Fribourg France have mortality rate of 102/100,000, yet the
cholesterol levels are identical at 245 mg/dl in both regions. The National Cholesterol Education Program is a quasi-governmental body
sponsored by members of the National Institutes of Health, American Heart
Association, and other supporters of the “diet-heart” hypothesis.
This body recommends a low fat, high carbohydrate diet to prevent
heart disease, in spite of the increasing incidence of diabetes, obesity,
and hypertension that is linked to consumers of this diet.
They consistently advocate programs of extreme lowering of
cholesterol levels by drug therapy, in spite of evidence of increased risk
of mortality from heart failure, cancer, cirrhosis, and other diseases in
older subjects with low cholesterol levels.
They also recently recommended lowering the acceptable level of Low
Density Lipoprotein (LDL) in the population by statin therapy, in spite of
the fact that 8 of the 9 members of the advisory panel had a direct conflict
of interest by accepting payments from the drug industry.
This body has popularized the concept that LDL is “bad cholesterol”
and HDL is “good cholesterol” in spite of the marginal and sometimes
contradictory data distinguishing these fractions from total blood
cholesterol. This body also
advocates “aggressive cholesterol lowering” in the population in spite
of the fact that no cholesterol lowering trials have demonstrated reduced
mortality or sudden death from such treatments in the otherwise normal
population. Homocysteine Homocysteine is an amino acid that is important in sulfur metabolism, as
discovered by the prominent American biochemist Vincent DuVigneaud in 1932.
He discovered that this amino acid has one more carbon atom than
cysteine, an important constituent of all proteins, and gave it the name
“homocysteine” [homo = same in Greek].
An amino acid called “cystic oxide” was discovered by Wollaston
in 1810 by isolation from bladder stones, and was later shown to contain
nitrogen by Berzelius in 1833, hence the name cystine [kystis = bladder in
Greek]. In working with
homocysteine and the amino acid methionine, DuVigneaud discovered its
importance in supporting growth of animals lacking methionine by chemical
transformations in the body called transmethylation. Little was known about the significance of homocysteine in human disease
until 1962, when cases of the new disease homocystinuria were discovered
among children with mental retardation, dislocated ocular lenses,
accelerated growth, osteoporosis and a tendency to form blood clots in
arteries and veins. Your
essayist became interested in the possible connection between homocysteine
and arteriosclerosis in 1968 through review of a case of homocystinuria from
1933 that was discovered by pediatricians at Massachusetts General Hospital.
This archival case was the uncle of a 9-year-old girl with mental
retardation, dislocated lenses, and abnormal blood vessels of the skin who
was diagnosed in 1965 by the new Amino Acid Laboratory directed by Mary
Efron and Vivian Shih. As
reported in the New England Journal of Medicine, the boy from 1933 died of
thrombosis of the carotid artery and a massive stroke, and the pathologist
Tracey Mallory found that the arteries were narrowed by arteriosclerosis
“similar to changes found in a very elderly man.”
Because of an interest in amino acid metabolism and experience with the
biochemistry of methionine and homocysteine at the National Institutes of
Health, I read the pertinent literature on this new disease, confirmed the
presence of arteriosclerosis in slides surviving from the 1933 case, and
found plaques scattered through the arteries of this child.
By chance I was able to study the case of a 2-month-old baby boy who
had recently died of the new disease, cobalamin C disease, characterized by
excretion of homocysteine, cystathionine, and methyl malonic acid in the
urine. Realizing that this case could shed light on the possible
connection between homocysteine and arteriosclerosis, I restudied the
tissues of this new case and discovered astonishingly advanced changes of
rapidly progressive arteriosclerosis. Since
the enzyme deficiency and the pattern of abnormal metabolism were different
from the archival case from 1933, I concluded that the amino acid
homocysteine produced arteriosclerosis in these children by a direct effect
on the cells and tissues of the arteries.
There was no evidence that lipids are deposited in the arteries of
these children, and no cholesterol was found in the arterial plaques. In experiments with rabbits utilizing similar a similar approach to that
of Ignatowsky and Anitschkow more than a half century earlier, my research
group at Massachusetts General Hospital discovered that administration of
the pure amino acid homocysteine to these animals produces arterial plaques
and thrombosis of veins and arteries. Simultaneously
giving pyridoxine (vitamin B6) to the animals prevents the plaques and
thrombosis. Although I did not
know it at the time these experiments were completed in 1975, Fumio Kuzuya
in Japan repeated our experiments and observed essentially the same results
in the late 1970s, reporting his reports in Japanese articles.
These results were also observed by Harker and Ross in Seattle,
utilizing intravenous homocysteine administration to baboons.
Thus these animal experiments led to the formulation of the
homocysteine theory of arteriosclerosis in 1975.
This theory helped to explain several important previous observations
of experimental plaques in monkeys deprived of vitamin B6, animals made
hypothyroid by thiouracil, and animals deprived of choline and other methyl
donor nutrients. Because the B vitamins folic acid, pyridoxine and cobalamin are all
involved in normal metabolism to prevent excessive production of
homocysteine, the homocysteine theory of arteriosclerosis implicated
deficiencies of these vitamins in human arteriosclerosis, heart attacks,
strokes and amputations from vascular disease.
Because the sensitive vitamins folic acid and pyridoxine are
destroyed by traditional methods of food processing, such as milling of
grains, canning, extraction of sugars and oils from whole foods, and
addition of chemical additives to bleach or preserve foods, this theory
helped to explain the large increase in vascular disease in the early and
mid 20th century. Similarly,
the decline in mortality from cardiovascular disease beginning in the 1950s
can be attributed to addition of the synthetic B vitamins to the food supply
by fortification of processed foods. Important
support for the homocysteine theory was provided by the Framingham Heart
Study, which showed that deficiencies of these B vitamins are widespread in
older participants, leading to elevation of homocysteine levels and
increased risk of plaques. Many hundreds of studies by investigators worldwide have now proven that
elevated homocysteine levels increase the risk for heart disease, stroke,
peripheral vascular disease, and reduce longevity in diverse populations.
In 1998 the US Food and Drug Administration mandated the addition of
folic acid to refined flour, rice and other grain foods, the first such
action since niacin, thiamin, riboflavin and iron were mandated for addition
to refined grain foods in 1941. One
of the rationales for adding folic acid is the demonstration that deficiency
of dietary folic acid is implicated in susceptibility to birth defects of
the neural tube type. An
additional rationale, not officially cited by the FDA, was the hope that
vascular disease incidence and complications might also be prevented in the
US. Studies have indeed shown
that birth defects have decreased about 19% in the US and up to 78% in
Canada in Newfoundland since fortification by folic acid.
In a recent study by the Centers for Disease Control and Prevention
in Atlanta, the declining incidence of mortality from stroke accelerated in
1998 in the US, but no change was found in the United Kingdom, where folic
acid fortification is not mandated. Last
year in 2007 a meta-analysis, as reported in Lancet, concluded that trials
yielding significant reduction in blood homocysteine levels from folic acid,
pyridoxine, and cobalamin over a 3 to 5 year period produced significant
reductions in mortality from stroke. Trials
with participants with a history of advanced vascular disease, heart attack
and stroke, have been less successful.
The Swiss Heart Study showed that restenosis following coronary
angioplasty was benefited by B vitamins, but a later trial with stented
patients showed no benefit. In the 1970s, before my removal from Harvard medicine, and in the 1980s
and 1990s my research groups at Massachusetts General Hospital and at the VA
Medical Center in Providence made some additional discoveries in the
homocysteine field. By using
cultured cells from children with homocystinuria, we were able to
demonstrate a new pathway for sulfur metabolism by showing that homocysteine
thiolactone, the reactive anhydride of homocysteine, is a precursor of
sulfate. We discovered that
this pathway is depressed in aging animals and completely blocked in
cultured cancer cells. Using
methods of organic synthesis, we developed and discovered new derivatives of
homocysteine thiolactone, retinoic acid, and cobalamin named thioretinamide
and thioretinaco. These
compounds are effective in preventing arterial plaques in animals treated
with homocysteine. Theorizing
that these compounds are deficient in cancer cells, we were able to decrease
cancer formation from chemical carcinogens and to decrease growth of
transplanted cancers in mice. These
compounds have not as yet been tested in human trials.
My second monograph describes a theoretical biochemical process by
which these compounds support the normal oxidative metabolism in normal
cells that is deficient in cancer cells. Infections
and vulnerable plaques We have learned that cholesterol plays a minor role in creation of
plaques, at least in younger men, and that homocysteine elevation increases
the formation of plaques and risk of disease in animals and in human
populations, through dietary deficiency of B vitamins, genetic factors, and
altered metabolism of methionine. However,
there are many other factors that are known to increase risk of vascular
disease. In particular, mental
and emotional stress, smoking, aging, male gender, postmenopausal status in
women, diabetes, kidney failure, hypothyroidism, and high blood pressure are
all important determinants of disease risk.
Surveys, case control studies, and experimental studies have shown
that each of these factors increases homocysteine levels in animals and man. How do these diverse factors conspire to produce
vulnerable plaques in the arteries that by rupturing cause hemorrhage,
thrombosis with occlusion, and death of heart, brain and other tissues in
patients with vascular disease? A century ago the cause of arteriosclerosis was generally considered to
originate from “direct irritation of [arterial] tissue by infection or
toxins.” Bacteria and viruses
were considered as the main cause of atherosclerosis, because of increased
plaques in patients dying of typhoid fever and other infections.
Sir William Osler, the Canadian pathologist and physician who became
the Regius Professor of Medicine at Oxford and a leading physician of his
time, described the vulnerable plaque as a pustule.
More recently, much evidence has been reported to support the role of
infections in vascular disease. In
particular, cardiovascular mortality increases during influenza epidemics.
A third of patients with acute myocardial infarction or stroke have
had an infectious disease immediately before onset.
Bacteriemia and periodontal infections are associated with an
increased risk of cardiovascular disease.
Serological markers of infection are elevated in patients with
cardiovascular disease. And
coronary arteries of children are found to be narrowed in children who died
of infectious diseases. Beginning in 1939 and in the next decade, investigators discovered that
lipoproteins function as a nonspecific immune system that is capable of
inactivating a wide variety of bacteria, viruses, protozoans and their
toxins. Lipoproteins form complexes with these organisms that render
them inactive, and macrophages in the tissues are capable of phagocytosis of
these complexes, leading to formation of foam cells that destroy these
organisms. The
lipopolysaccharides and lipoteichoic acids of these organisms bind with
lipoproteins, probably because of their lipophilic properties, rendering
them inactive. Some examples of
organisms that are inactivated this way are Salmonella,
the cause of typhoid fever and other gastrointestinal infections, Herpes
simplex, Rotavirus and Cytomegalovirus,
the cause of ubiquitous viral infections, Chlamydia
pneumoniae, a respiratory pathogen,
and Staphylococcus aureus, an
important cause of boils, carbuncles, and pneumonia. Beginning in the 1970s investigators found evidence of a wide variety of
micro-organisms in human atherosclerotic plaques, as demonstrated by
immunohistochemistry, polymerase chain reaction for DNA fragments, and
electron microscopy. Only in
the case of Chlamydia pneumoniae were investigators able to culture viable
organisms directly from plaques. In
most cases, only remnants of micro-organisms are found, and viable organisms
cannot be cultured from plaques. Using
sophisticated DNA technology, a recent report from Germany demonstrated the
presence of 50 different micro-organisms in human plaques, and the average
number was 12 per plaque. Arteries
without plaques contained no microbial remnants. As shown in the figure, many common organisms form complexes
with lipoproteins, such as Escherichia
coli, Staphylococcus, Streptococcus, Salmonella, and rotavirus.
The same organisms are demonstrated in plaques, suggesting an
infectious origin of plaques. Homocysteine participates in the aggregation of lipoproteins by reacting
with the protein to form cross-linkages, leading to aggregation, spontaneous
precipitation and phagocytosis by macrophages to form foam cells. Thus in persons with elevated homocysteine levels, there is
an increased ability of lipoproteins to form aggregates, leading to foam
cell formation from macrophages by phagocytosis.
In addition, lipoproteins containing homocysteine have altered
antigenic structure, leading to the formation of antibodies that complex
with lipoproteins. A similar
process has been identified with lipoproteins within foam cells, since
oxidation reactions create modified or oxidized lipoproteins that incite
autoantibody formation. In
the presence of invading micro-organisms, the complexes with lipoproteins
theoretically become enlarged and subject to precipitation and phagocytosis
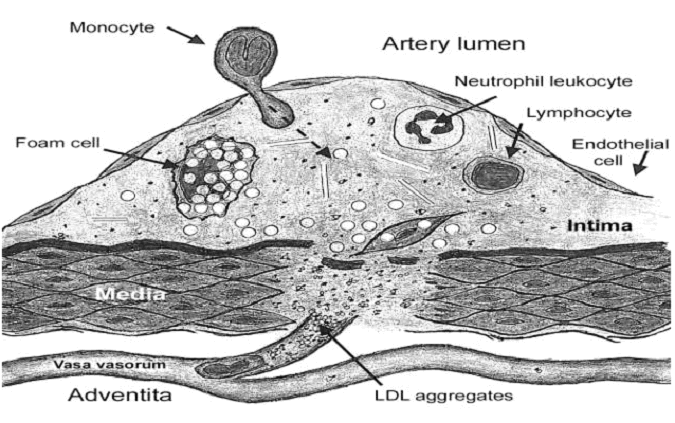
by macrophages. The figure illustrates our concept of the creation of vulnerable plaques within arteries. In the case of a massive invasion of micro-organisms, or when the immune system is impaired, the complexes of micro-organisms with lipoproteins, enhanced by the effect of excess homocysteine, obstruct the vasa vasorum, the small arterioles, venules and capillaries that normally nourish the wall of the artery. Obstruction of the blood flow causes ischemia of the artery wall (lack of blood supply), increasing the tendency to cell death and rupture of the capillaries. These large complexes are phagocytosed by macrophages to form foam cells that accumulate, initially in the adventitia, but then migrating into the intima, where lipoproteins, inflammatory cells, and lipid deposits, including cholesterol, accumulate. This process is probably increased and enhanced in cases where autoantibodies are formed to homocysteinylated lipoproteins and oxidized lipoproteins. This process is further exacerbated by swelling of endothelial cells and narrowing of the lumens of vasa vasorum by endothelial dysfunction, caused by elevated homocysteine and other factors. The result is a vulnerable plaque that by rupturing causes thrombosis, obstruction of the arterial lumen, and death of the heart, brain, kidney or other tissue supplied by the artery. The vasa vasorum are functionally end arteries, since blood flow ceases at the point where the pressure within these blood vessels is opposed by the pressure within the arterial lumen. Thus, the process of plaque formation is potentially enhanced in areas of increased blood pressure, such as disrupted laminar flow, turbulence, angulation of arterial lumen, and increased systemic pressure. In fact, these are the areas in the arteries where vulnerable plaques are most commonly observed.
Fig.
1. Development of the vulnerable plaque. The small globules inside the vasa
vasorum and in the vulnerable plaque represent lipoproteins; the black dots
represent microorganisms, ndotoxins, anti-OxLDL autoantibodies, and
anti-thiolated-LDL autoantibodies; the large globules at the basal part of
the vulnerable plaque and inside the macrophages represent lipid droplets.
The right capillary represents the situation in a normal healthy artery;
there are only a few microbes and the lipoproteins are able to traverse the
capillary lumen without adherence or obstruction. The left capillary
represents the situation in an artery with a severe microbial invasion;
microbial products and autoantibodies stick to the lipoproteins, which
aggregate and obstruct the capillary lumen, leading to local ischemia,
microbial growth, and inflammation. A monocyte enters the plaque from the
arterial lumen by diapedesis between endothelial cells; another monocyte
enters the plaque via vasa vasorum, leading to formation of foam cell
macrophages within the plaque. In the case of an intact immune system, the
inflammatory area heals and becomes converted to a fibrous plaque. In the
case of an insufficient immune system, microorganisms escape into the tissue
and create a microabscess, the vulnerable plaque. (From Ravnskov U,McCully
KS. Ann Clin Lab Sci 2009;39:3-16. One can readily understand that our interpretation explains many of the
observations about vulnerable plaques and the pathophysiological processes
found in vascular disease.
It explains how risk factors, such as stress, B vitamin deficiency,
elevated blood pressure, smoking, and kidney failure lead to increased
disease risk through formation of homocysteinylated lipoprotein aggregates.
It explains the resistance of persons with elevated lipoprotein
levels to infectious diseases.
It explains how cholesterol becomes deposited in plaques.
It explains the inflammatory process and the release of inflammatory
cytokines, C-reactive protein, fever, leukocytosis, and the frequent
occurrence of bacteriemia and sepsis in myocardial infarction complicated by
shock. It
explains the observation of microbial remnants in plaques.
And it explains the inflammatory nature of cardiovascular disease. Suggestions for prevention and therapy of vascular disease readily
follow from our interpretation of the origin of vulnerable plaques.
Risk factors that lead to elevated homocysteine levels need to be
addressed by smoking cessation, dietary B vitamins, stress reduction, and
control of diabetes and hypertension. The
immune system needs to be enhanced by intake of vitamin A, vitamin D,
pyridoxine, and other factors that are needed for proper immune function.
Appropriate antibiotic therapy is needed in heart attack and stroke
patients that have evidence of active infection.
Dietary improvement is needed to supply the nutrients that are needed
to minimize homocysteine elevation and to support anti-oxidant function.
Attempts to prevent cardiovascular disease and prolong life may be
more successful by understanding the fallacies of the “diet-heart”
hypothesis and determining what is harmful to the immune system and what may
strengthen it. Sources Keys A. Coronary heart
disease – The global picture. Atherosclerosis
1975;22:149-192. McCully KS. Hyperhomocysteinemia
and arteriosclerosis: historical
perspectives. Clin
Chem Lab Med 2005;43:980-986. Ravnskov U, McCully KS. Vulnerable plaque formation from obstruction of vasa vasorum by homocysteinylated and oxidized lipoprotein
aggregates complexed with microbial remnants and LDL autoantibodies.
Ann Clin Lab Sci 2009;39:3-16. Ravnskov U. The Cholesterol
Myths. Exposing the fallacy
that saturated fat and cholesterol cause heart disease. New Trends Publishing, Washington DC, 2000. Levy D, Brink S. A Change
of Heart. How the people of
Framingham, Massachusetts helped to unravel the mysteries of cardiovascular
disease. Knopf, New York, 2005. McCully KS. The
Homocysteine Revolution: Medicine
for the New Millennium. Keats
Publishing, New Canaan CT, 1997. McCully KS, McCully ME. The
Heart Revolution. HarperCollins,
New York, 1999. McCully KS. Homocysteine,
vitamins and vascular disease prevention.
Am J Clin Nutr 2007;86:1563S-8S. Ravnskov U. Fats and
cholesterol are good for you! GB
Publishing, Sweden, 2009. Peskin BS, Sim D, Carter MJ. The
failure of Vytorin and statins to improve cardiovascular health:
bad cholesterol or bad theory? J
Am Phys Surg 2008;13:82-87. Kastelein JJP, Akdim F, Stroess ESG.
Simvistatin with or without ezetimibe in familial
hypercholesterolemia. New Eng J
Med 2008;358:1431-1443. McCully KS. The Chemical
Pathology of Homocysteine. IV.
Excitotoxicity, Oxidative Stress, Endothelial Dysfunction, and
Inflammation. Ann Clin Lab Sci
2009;39:307-320.
|