|
Top
Nicolai
Worm
This
is something to think about:
J Am Coll Cardiol. 2005 Jun 7;45(11):1794-801.
Miettinen TA, Railo M, Lepantalo M, Gylling H.
Plant
sterols in serum and in atherosclerotic plaques
of patients undergoing carotid endarterectomy. Department
of Medicine, Division of Internal Medicine,
University of Helsinki, Helsinki, Finland.
OBJECTIVES: The purpose of this research
was to determine whether serum plant sterol
levels are associated with those in atheromatous
plaque.
BACKGROUND: Cholesterol of low-density
lipoprotein (LDL) particles contributes to
atheromatous plaque formation; LDL also contains
most serum non-cholesterol sterols, including
plant sterols. The role of plant sterols in
atheromatous plaque formation is open. METHODS:
Free, ester, and total cholesterol and the
respective non-cholesterol sterols were measured
by gas-liquid chromatography in serum and
arterial tissue of 25 consecutive patients
undergoing carotid endarterectomy. The
population was ranked to triads according to
tissue cholesterol concentration.
RESULTS: Cholesterol concentration
increased markedly in tissues but not in serum
with triads. The ester percentage was lower in
the third than in the first triad (47% vs. 56%;
p < 0.01) and lower than in serum triads
(70%; p < 0.001). Ratios to cholesterol of
non-cholesterol sterols decreased in increasing
tissue triads, but were unchanged in serum. A
major new observation was that the higher the
ratio to cholesterol of the surrogate absorption
sterols (cholestanol, campesterol, sitosterol,
and avenasterol) in serum, the higher was their
ratio also in the carotid artery wall (e.g., r =
0.683 for campesterol). Despite undetectable
differences in serum and tissue cholesterol
concentrations off and on statins, an additional
important novel finding was that statin
treatment was associated with increased ratios
of the absorption sterols in serum and also in
the arterial plaque.
CONCLUSIONS: The higher the absorption of
cholesterol, the higher are the plant sterol
contents in serum resulting also in their higher
contents in atherosclerotic plaque. However, the
role of dietary plant sterols in the development
of atherosclerotic plaque is not known
Top
Leib
Krut
It
is amazing how things are rediscovered. I do not
have references at my fingertips, but I do
recall that several decades (~4) ago there was
interest in feeding plant sterols with the idea
that these would compete with cholesterol for
absorption. This did indeed prove to be the
case. Unfortunately the hopes inherent in the
concept that lowering cholesterol in plasma is
beneficial were dashed when it was found that
feeding these plant sterols increased the plant
sterol content in arteries. It was not possible
to know what the relevance of this was, but it
did not seem to be a plus, and, so far as I
know, there was no more exploration of this
topic, until now. I think Miettinen should be
aware of that work done decades ago, although he
might need to dig deep into his memory bank to
recall those studies. ( I have not seen their
paper, and they might well quote the earlier
work. I shall look it up). What this work does
indicate is that sterols, (and possibly other
compounds) contained in LDL do become trapped in
the arterial wall when LDL is trapped there. The
crucial issue is: How is this relevant to our
problem?
Now
that I am scratching my own memory bank, I
recall that Merck marketed a compound named Mer
29 about 1960 (I think), which blocked
cholesterol synthesis at or near its penultimate
stage. There was an accumulation of that
precursor(s) in the plasma, with lowering of
cholesterol, but that precursor also found its
(their) way into arteries. I do not remember
whether these precursors were also shown to be
transported in LDL, but it seems likely they
were so transported. There were a number of
other deleterious effects of Mer 29 thought to
be alarming at the time and it was dropped (it
might have survived in todays climate!). This
experience would seem to have been the reason
for the search for a compound that would block
cholesterol synthesis early in its synthetic
pathway. And so we acquired statins! Like many
of you, I expect that there will ultimately be a
high price to pay for blocking cholesterol
synthesis early in its synthetic pathway.
It
would seem to me that those who believe that
cholesterol from plasma has relevance in
atherogenesis need to establish what it is that
converts it into a pathogenic moiety in the
artery, how that might be prevented, and the
relevance of such prevention in the epidemiology
of CHD. Surely we are by now beyond the stage of
putting all our focus on plasma cholesterol
concentration and how to lower that
concentration by every conceivable means.
Top
Morley
Sutter
You
are absoulutely right about Mer 29 except that
it was not Merck, but a company called Merrell
that marketed it.
Mer 29 was taken off the market because
it caused cataracts.
The suits almost broke Merck. Merrell
subsequently disappeared in some merger or
other.
Top
Barry
Groves
And
you don't have to go back 40 years. See Plat J,
Brzezinka H, Lutjohann D, Mensink RP, von
Bergmann K. Oxidized plant sterols in human
serum and lipid infusions as measured by
combined gas-liquid chromatography-mass
spectrometry. J Lipid Res 2001
Dec;42(12):2030-2038.
Dr Plat and colleagues at Maastricht
University’s Department of Human Biology in
the Netherlands, say that plant sterols may
actually be more important in heart disease than
cholesterol.
Cholesterol
is only thought to be harmful if it is oxidised.
Because plant sterols are structurally related
to cholesterol, Plat and colleagues examined
whether oxidized plant sterols (oxyphytosterols)
could be identified in human blood and
soya-based fat emulsions. They found that they
could. Approximately 1.4% of the plant sterol,
Sitosterol, in blood was oxidised. That's 140
times as much as the 0.01% oxidatively modified
cholesterol normally seen in human blood. They
found the same with two soya emulsions.
Top
Leib
Krut
Thanks
for your input. It did come back to me. The
compound that accumulated with Mer 29 was
Desmosterol and it was found in plaques and the
drug did cause cataracts.
The
work of Miettinen et al shows that a number of
cholesterol precursors are found in plasma and
plaques. I suspect they may be implying too
much. It seems likely that these precursors (and
plant sterols derived from the diet) are
transported in LDL and trapped with LDL in the
subendothelial space of arteries. I would
suggest that their findings in plaque do not
reflect recent depositions. The lipid in plaque
is pretty well encapsulated by fibrous tissue.
There is usually no lipid in the fibrous
capsule. The lipid in plaque is contained in
"pultaceous necrotic tissue",
according to pathologists. It is not part of a
metabolic pool. This lipid must for the most
part, if not entirely, be laid down early in
life. It is most unlikely that its content or
composition could be changed by more recent
alterations in plasma, either by reductions in
plasma cholesterol concentration, eg with
statins, or by induced alterations in the
concentration of other sterols in plasma.
The
concentrations of cholesterol precursors in
plasma and of plant sterols that Miettinen et al
measured are an exceedingly small fraction of
the total sterols in plasma, the great bulk is
cholesterol. We have no idea what the presence
of these other sterols means; probably nothing
of note, tho' there are a number of other
aspects one may infer from that study which are
of more general interest.
Dear
Barry: Thank you for your input and for the
reference to Plat et al on plant sterols. My
point really was that the possible role of plant
sterols in atherogenesis is a very old idea
which had not lead to any contribution to our
understanding of what that disease process is
about. This does not mean that it is not
deserving of another look. However, just
considering concentrations of compounds of
interest does not seem to have lead us anywhere
except into what most THINCS members agree is a
conceptual morass.
I
am interested in the points you make about
oxidized phytosterols. I think expressing their
concentration the way you have done does put a
slant on things. To say that the amount of
oxidized phytosterol is 140 times the amount of
oxidized cholesterol will seem alarming to those
who believe that oxidized cholesterol, perhaps
other sterols, is/are the lethal compound/s.
However, the fact that 1.4% of phytosterol is
oxidized compared with 0.01% of cholesterol that
is oxidized should not be alarming. It can be
noted in the Miettinen paper that the amount of
phytosterol in plasma or tissue relative to the
amount of cholesterol is minute. 1.4% of
oxidized phytosterol leaves a minuscule amount
of these compounds in plasma. There is no
possible way these negligible quantities of
oxidized sterols could have relevance in a
process such as atherogenesis. Such minute
quantities of sterols might conceivably have
relevance if they had hormonal implications. We
all know that a huge number of factors have been
implicated in atherogenesis and related issues.
On reflection it is perhaps surprising that no
one has as yet (at least to my knowledge)
implicated a steroid hormone, conceivably
derived from ingested sterols (!), in its
genesis. Perhaps that will still be done down
the road; anything goes in this field.
I
might add that a possible reason for such minute
quantities of oxysterols in plasma is
attributable to the extraordinarily rapid rate
at which oxysterols are cleared from plasma
relative to cholesterol. (Krut
et al. Arterioscler
Thromb Vasc Biol. 1997;17:778-785, Correction
1979;17:1481) The actual amount traversing
plasma might be more substantial.
Raising
the spectre of oxidized cholesterol does give me
the opportunity to plug my views. Oxysterols, as
distinct from cholesterol, were first
conceptually implicated in experimental
atherogenesis by Altschul around 1946. Since
then it has over the years been reported by
several groups of workers, including work by
Altschul, that oxysterols do the reverse. They
in fact attenuate lesion formation in
cholesterol fed animals. The work implicating
oxysterols in atherogenesis would seem to be
questionable.
I
have postulated that cholesterol from plasma
becomes a pathogenic moiety in the arterial wall
when it takes on its native character, which is
that of a crystalline solid. In this state
cholesterol cannot be cleared from tissue and it
is sclerogenic. Thus factors that promote or
prevent the crystallization of cholesterol
determine its role in atherogenesis. This view
is based on observations originally made by
chance, namely, that oxysterols prevent the
crystallization of cholesterol in in vitro
systems, and that glucose promotes the
crystallization of cholesterol. In addition,
oxysterols implanted subcutaneously in rats
together with cholesterol results in the
solubilization and clearance of a large mass of
cholesterol, leaving little residual fibrosis.
Implanted pure cholesterol is rapidly
sequestrated by fibrous tissue, no cholesterol
is cleared and it remains sequestrated in a
seemingly permanent granuloma.
The
possible relevance of oxysterols to
atherogenesis in humans was attributed to their
progressive elimination from the human diet
since the advent of refrigeration, which
prevents the spontaneous oxidation of
cholesterol in foods of animal origin.
Oxysterols must have been invariably generated
in such foods by the techniques practiced for
preservation of these foods prior to the
development of refrigeration. Oxysterols would
have been progressively eliminated from the
human diet with progressive application of
modern technology in preservation and handling
of foods.
You
can find references, should you want to follow
up on these matters, in: Krut L H. Med.
Hypotheses 1979;5:533-548, Atherosclerosis
1982:43;95-104 and 1982:43; 105-118. Recent Res.
Devel. in Lipids Res 2, 1998:299-318. Amer J
Cardiol 1998:81;1045-1046. Atherosclerosis
supplements 2004;5/1:36. Wilkens and Krut. J.
Atheroscler Res 1963:3:15-23 and 1965:5;516-523.
Top
Bogdan
Sikorski
Leib
- When some months ago, during your introduction
to the group, you described your research
hypothesis about the positive role of oxchol in
atherosclerosis, I could "feel" the
unease it created - no one, including me, dared
to question you on that. Perhaps
"dared" is a wrong term, but certainly
the typical skepticism of the group was
surprisingly dormant.
Now, what you have just again described really
makes sense to me (I hope to stir some unease
here) considering that I am of the firm belief,
or rather conviction, that circulating chol can
not have a major influence, if at all, on the
pathogenesis of atherosclerosis or formation of
the plaque. Whatever the precise mechanism is,
if there is just one, the in situ production
appears logically to be the major source of chol
with a major influence of glucose and insulin,
and possibly other factors, all in response to
an injury or anoxic assault of some form. This
fits in with your notion about the positive role
of oxchol in mopping up the injury site in an
attempt to restore the functional integrity of
the blood vessel. The putative role of glucose
and insulin on chol formation in situ has been
first documented by Stout in early 1970s using
radioligands.
I might add that another experiment which showed
that chol can be made in situ, in media, before
the arrival of macrophages, subject to anoxic
assault, has been done on aortae of living
rabbits (this time I am confident also without
feeding chol) fitted with inflatable cuffs,
after separation from vasae vasorum. I do not
know if these results were ever published, since
I heard them reported at a scientific meeting
some year ago. I forgot how they differentiated
between the in situ mechanism and infiltration
of chol from the lumen, but I remember that I
asked the experimenter if they used statins to
block the in situ synthesis - they did not at
the time, but planned to do so.
Anyway, contrary to popular modern dietary
habits, I and my family must be just about the
only human beings, apart from the remnants of
traditional Northern Indians, who are happy to
consume buckets of ghee, which as a rule must be
relatively high in oxchol. We (well, my wife)
make our own, to which we also add some coconut
oil, which makes it a perfect frying, non
smoking, fat.
Oh well, this week for a change we will have a
bucket of freshly melted lard, after procuring
some non-smelly pork fat from a Chinese source.
Hopefully, that should also be oxchol rich due
to the high-temp melting process.
Here is hoping for clear arteries!
Top
Alena
Langsjoen
Dear
Jacqueline, Zetia (Ezetrol or Ezetimibe) is a
pretty strange looking molecule which somehow is
supposed to block absorption of dietary
cholesterol along the brush border of the small
bowel. Here in the US it is marketed by itself
and also in combination with Zocor (this
combination drug is called Vytorin). Peter has
been worried about Zetia and has not prescribed
it to any of his patients. But some of his
patients are advised by their primary doctors to
take it, or worse yet, to take Vytorin which
obviously is a double whammy.
If
Zetia blocks the absorption of cholesterol, does
it also block absorption of dietary fat-soluble
vitamins, like vitamins D, E, betacarotene and
lycopene, essential fatty acids and also how
about our favorite molecule, the fat-soluble
CoQ10-some of which we get from our diet? The
PDR states that (this is not an exact quote)
after 14 days supplementation with Zetia, there
is no clinically significant depletion of fat
soluble vitamins. Peter and I are now able to
analyze patient plasma in our laboratory with
our new HPLC system equipped with
electrochemical detector (ECD) and UV detector
(UVD). Our ECD produces a graph with peaks for
vitamin E, lycopene, beta carotene, reduced
CoQ10 (H2CoQ10 or ubiquinol) and oxidized CoQ10
(ubiquinone). Our UVD produces graph with peaks
for free cholesterol and several cholesteryl
esters (linolenate, arachidonate, linoleate and
oleate).
Even
though I tried objecting to Peter doing this (I
like for Peter to stay healthy) towards the end
of last year Peter decided to do an experiment
on himself by taking Zetia for 2 weeks. We ran
his baseline plasma and again after 2 weeks on
Zetia. FYI, after I post this I will upload pdf
of the resulting analysis of his plasma. First
page of the pdf is his plasma while he was
taking 600mg CoQ10/day for at least a month (no
other supplements). The second page is his
plasma after 2 weeks on Zetia in addition to the
600mg CoQ10/day. Two weeks was all he could
stand taking the Zetia. Note that his total Q10
(oxidized + reduced Q) level dropped by 43%. And
his vitamin E level dropped by 33%.
I
cannot quantify the lycopene or beta carotene
peaks because I don't have standards for them
yet, but after 2 weeks on Zetia Peter's area
under the peak for lycopene dropped by 26% and
for beta carotene by 48%. It is noteworthy that
the cholesteryl esters also dropped, along with
the free cholesterol. We do not know how this
relates to the status of plasma levels of free
fatty acids. Note the total Q10/total
cholesterol ratio dropped (by 18%).
Next
we thought it would be very interesting to find
out what effect Zetia has on people who are not
supplemented with CoQ10 so we are recruiting
some volunteer doctors and we are in the process
of collecting
data. We plan to publish the results.
Top
Bogdan
Sikorski
Alena
- This is very interesting. I hope I can show
these HPLC results to few people, but a
published study would of course be very powerful
indeed. In AUS, the dietary use of phytosterols
has been thus far restricted (because of effects
on fat-sol Vits) to "healthy"
margarine, but the "good" industry has
been pushing to have these timber milling
byproducts (i.e. normally environmental
pollutants) to be included in a wide range of
products, including kids "health" bars
and milk-drinks for some time. I think, the US
FDA being more "progressive" has
already allowed a relatively wide use of
phytosterols in foods such as milk and soft
drinks. Treatment with these could be another
arm of your study after a 2-week washout period.
Top
Alena
Langsjoen
We hope to publish our Zetia findings, but it
may be a while before we get to that. I'll see
if I can talk Peter into eating a bunch of
phytosterols for an experiment. Actually right
now we really want to concentrate on measuring
tissue levels of CoQ10 and correlating that to
plasma levels. We hope to start doing this
sometime this fall or winter. A local
cardiovascular surgeon is willing to cooperate
with us, giving us a snip of the heart along
with a concurrent blood sample.
Top
Eddie
Vos
Alena,
It is wonderful that after blowing the kids'
college fund to a chromatograph it is finally
spitting out reproducible results!
You did not say what dose zetia Peter was
on, I presume 10 mg and where the drop in total
cholesterol reported in the P.I.., below, is
more like -14%, your finding was -30%
.. http://www.zetia.com/zetia/shared/documents/zetia_pi.pdf
Other links:
http://www.vytorin.com/ezetimibe_simvastatin/vytorin/hcp/price_dosing/dosing.jsp
http://www.zetia.com/ezetimibe/zetia/hcp/index.jsp
It
is also amazing to me that cholesterol in serum
marries so massively to linoleic acid [excess
n-6 in the diet?] and also that the on ezetimibe
test showed n-3 linolenate cholesterol ester
dropped from 0.04 to 0.01 mMol, but I guess that
could be limit of detection, or less
alpha-linolenic acid in the diet the day before
since it does not store for long.
Final
comment: I would not dream about going a day
without a high dose multivitamin to keep my
homocysteine at a low level; Peter being on no
other supplement than Q10 sounds like he's
missing something, but that's the subject of a
next email. Best
to you both and exiting stuff that new toy!
Top
Alena
Langsjoen
Peter's Zetia dose was 10mg/day.
Yes,
we're having lots of fun with our new expensive
HPLC toy after some stressful times with it.
I agree that the n-3 linolenate chol.
ester is very close to detection limit, not only
because its concentration is so low but also the
peak is not very well separated from cholesteryl
arachidonate which comes off right after the
chol.linolenate. I got a big bottle of B-50
complex from Sam's on Peter's side next to his
sink in our bathroom now and I try to remind him
to take one every day.
Top
Melchior
Meijer
What a creative and courageous
experiment. I have some questions. Zetia
(Ezetrol) blocks (re-)absorption of cholesterol
from the gut and has (unlike statins) no
influence on endogenous Co Q10 synthesis. The
fall in Q10 in Peter's plasma is thus a
reflection of Zetia blocking the uptake of the
Q10 supplement and Q10 is his food, right?
- Are healthy
individuals depending on dietary Co Q10 and if
so, to what extend?
- Do indivuals
that habitually supplement with large doses of
Q10 somehow lose their ability to synthesise
their own Q10, or do you think Peter's side
effects were merely due to lower cholesterol
(BTW, what exactely did he feel)?
- Would Peter be
willing to 'test' Becel Pro Aktiv (starring some
potentially nasty plant sterols) in the same
way? As I reported earlier, Dutch 'patients'
with a TC of > 5 mmol/L get their Becel Pro
Aktiv paid for by their health insurance. This
is a big, sick, scandalous joke, perpetuated on
TV by our Heart Association. Especially in an
already n-6 overfed population, this n-6 laden
stuff is insulinotrophic, pro-infammatory,
angiogenic, carcinogenic and probably
atherogenic (Simopoulos, Enig, Ottoboni, Okuyama
et al). If it also turns out to mess with the
uptake of important nutrients (especially A and
D, which are added!), it could make headlines.
Top
Alena
Langsjoen
Yes, we think that Zetia blocked the
absorption of Peter's large doses of
supplemental CoQ10 and it also blocked the
ability to carry Q10 (Q10 gets carried by LDL
& VLDL).
At 600mg/day you could say that CoQ10
could almost be Peter's food but he still has a
good appetite for eggs, butter, steak and all of
my Czech cooking!
Some
of our current observations are making us
believe that dietary CoQ10 may be much more
important than previously thought.
Nakamura
has performed experiment with radioactively
labelled supplemental CoQ10 in animals and there
was no decrease in endogenous biosynthesis of
CoQ10. I
can find the reference if you need It.
Peter's
side effects were weakness, fatigue and some GI
distress. Peter said that he would think about
experimenting with Becel Pro Aktiv.
Top
Leib
Krut
Dear
Bogdan: Thank you for your note. I must say I
was quite tickled to read of the reaction you
felt occurred in the THINCS group in response to
my concepts. I am not unaware of the general
reaction to my "heterodoxy". I must
say that I had hoped that in a group of
"heterodox" thinkers there might have
been more interchange with another view that was
very different from the mainstream.
I
moved to the USA about a decade ago and my
reprints of articles got into a mess, beyond
recovery. I mention this because I should have
liked to give you a reference to a paper
published in the British Heart Journal many
years ago by someone named Malhotra (I think).
He wrote on the difference in the incidence of
Coronary Heart Disease between Northern and
Southern Indians in India. He reported
that
the southerners were vegetarian and lean and had
a CHD incidence 7+ times that of the
northerners who were much heavier and who ate a
considerable amount of fat (10-20 times that in
the south), that this fat is mainly from animal
sources, including ghee, and therefore largely
saturated. In the south the small amount of fats
are mainly from seed oils and therefore largely
polyunsaturated.He described the way ghee was
made, something your wife clearly knows well,
and I have no doubt that oxysterols are
generated in the process since they are found in
butter made by traditional methods in which
cream is allowed to "ripen" simply by
holding it at ambient temperature for 3-4 days.
This cream is then churned to make butter. I
suspect that in making ghee even more oxysterols
are generated. I might add that the northerners
smoke 8 times as many cigarettes as do their
southern neighbours. They would seem to be doing
all the wrong things in the north, according to
the standard dogma, and yet are protected from
CHD, save for the fact that must have a
considerable intake of oxysterols, or at least
must have done so when Malhotra wrote that
paper. It goes without saying that I found that
paper very appealing and that clearly is why I
recall it so many years after its publication.
It may be that I cited it in one of my
publications. If I do come across it I shall let
you have the reference. As a potential ally, I
shall dearly love to provide you with as much
information as possible to encourage your
support.
I
did in fact cite that paper. The reference is:
Malhotra SL. Brit. Heart J. 1967;29:895-905.
I
think you will find it of interest.
Top
Barry
Groves
I
have a paper of Malhotra's on this subject, but
published in Am J Clin Nutr (Malhotra
SL. Serum lipids, dietary factors and ischemic
heart disease. Am
J Clin Nutr
1967; 20: 462-474). In
it Malhotra states " . . .occurrence rates
of acute myocardial infarction were seven times
higher in the South Indians as compared to the
North Indians, even though the North Indians
consumed nine times more fat, most of which was
animal fat derived from milk and ghee, with a
preponderance of saturated fatty acids" He
gives as a reference for this statement his own
paper, Geographical aspects of acute myocardial
infarction in India, with special reference to
the pattern of diet and eating. Brit. Heart
J. 1967;29:777.
Malhotra notes that in the comparison between
the two groups, serum cholesterol levels were
similar and normal values. Free fatty acids were
non-significantly higher in the Northern
Indians, but mean values for esterified fatty
acids, cholesterol esters and total
triglycerides were similar in both groups.
What this study shows is that serum lipid levels
can be the same and normal in peoples with very
big differences in both their intakes of fats,
including animal fats, and incidences of IHD. It
also demonstrates that serum lipid levels are
not dependent on totals or even proportions
between different fats eaten.
Exercise and physical activity are also
considered by Malhotra, but he found no
significant difference between the two groups --
they both were "habituated to an identical
amount of a high degree of physical exercise at
work . . ."
After measuring fecal and urine urobilinogen
excretions, he puts the blame for the South's
higher CHD rates on excess bile entering the
intestinal lumen. "These results . . . show
unequivocally that out South Indian group on a
carbohydrate-rich, lipid-poor regime of boiled
rice and lentil soups had significantly larger
amounts of bile in their intestinal lumen, as
compared with our North Indian group on
fat-roughage and cellulose-rich wheat diet; and
that these differences are dependent upon the
pattern of diet and eating."
Malhotra also talks about other studies that
buck the trend -- Bantu, Masai and Samburu in
Africa.
Top
Bogdan
Sikorski
I recall, that we have discussed this paper, or
rather these findings, in the past, on more than
one occasion.
I think, I looked for the journal in the
departmental (work) library and could not find
it. It would be nice to get a copy (scan?) of
both.
Anyway, Leib, I can hardly be your ally,
considering that I am "running on
empty" - I have done no research (except
for a dietary trial we are running at home) in
the area, but I am very interested in your
findings and the "fringe" or heterodox
hypothesis you have constructed based on them.
Ultimately, it would be nice to know that our
dietary preferences have some scientific
support.
As far as I can tell, many members of this group
are on a range of fringes, having adopted one or
more heterodox positions, with one uniting us
all. But, as with any group of people, one has
to be aware of not pushing too hard! In such
cases, the polite response is typically no
response or no comment.
Being brought up Central European, I don't have
much respect for such politeness - thus my
previous comment. Lets "drill" this
hypothesis of yours. Perhaps Uffe or Malcolm
should start? Ooops - maybe someone less
"polite" first? Did I offend anyone?
Top
Uffe
Ravnskov
First a comment to Bogdan. Hopefully no one in
this group who disagrees with thoughts presented
in our correspondence would remain quiet because
of politeness. I think not, because previous
discussions bear witness to many divergent
thoughts about this and that and hopefully we
shall proceed that way.
As
for Leib´s idea about oxidized cholesterol I
have speculated much about it without coming up
with any wise thoughts, mainly because my
biochemical knowledge is too rudimentary.
Personally I think that the idea about oxchol
being the villain is a typical ad hoc
hypothesis. As far as I know there is no
evidence of that besides the finding that high
oxchol is a better predictor than high
cholesterol, but there are probably more than
hundred other risk factors that are better
predictors than high cholesterol so why just
oxchol.
I
recently read The
Dynamics of Atherosclerosis (Aberdeen University
Press, Aberdeen 1976), a most interesting book
by British pathologist Jack Duguid. His
hypothesis is that atherosclerosis is the
sequels of microthrombi formed at the intimal
surface. Very quickly such thrombi are covered
with endothelial cells. Their repeated
incorporation into the intima throughout life
beginning in early childhood eventually results
in irregular fibrous thickenings that are too
stiff to comply wih pulse movements and so cause
disruption and haemorrhage. The extravasated
blood disintegrates leaving fatty deposits which
accumulate progressively. Thus, plaques are
simply scars and he
has illustrated his idea with many compelling
microphotographs.
The
crucial question is of course, what is causing
these microthrombi? Here is room for many
suggestions, for instance too much homocysteine,
free radicals, microorganisms, etc and none of
them exclude any of the others; a complex
interaction is likely.
The
finding of plant sterols in atherosclerotic
plaques is amusing but didn’t surprise me at
all. It is amusing because of the authors´
conclusion. If they think that high cholesterol
leads to atherosclerosis, which they obviously
do, then why don’t they follow the line and
warn against plant sterols? Or have they
realised that the finding of a certain substance
in a scar doesn’t mean that the same substance
is causing the scar?
Top
Paul
de Groot
Reading the discussions on atherosclerosis
(2002-2005), my main interest, some statements
are still relevant. I like to comment on two of
them. Firstly: intimal enlargement is the first
sign of atherosclerosis and secondly:
atherosclerotic plaques seem randomly
distributed over the arterial wall apart from
the branching sites.
My
studies of human coronary arteries contradict
the first statement while I endorse the second
one.
I
tell you my story and will try to formulate the
way of thinking.
1984
I planned my thesis study on human coronary
artery vasa vasorum. The heart foundation
was sceptical about even the existence of
vasa vasorum in the coronary arterial wall. The
intention was to study the normal coronary
artery and the artery circumferentially
enlarged, then called hypertrophied. Mind you,
in literature, no definition of “normal”
could be found and so also not of concentrically
enlarged vessels. Of course some histological
indications could be found but no hard figures
at all. We came up with the following
histological classification:
Coronary
artery N
AN A
E
INTIMA
Endothelium undamaged
undamaged undamaged
damaged
Sub-endothelial
Layers some
several many
many
Thickness
Intima/media <1/2
media in
between >1/2
media >>1/2media
Conc.c.q. ecc. -
conc. Conc.
Ecc.
Smooth
Internal
Elastic Lamina
Single/double single
single/double
double double
Continuous/Fragmented
cont. cont.
fragmented
fragmented
Undulated/Stretched/
Destroyed undulated
stretched stretched
destroyed
Extenal
Elastic Lamina
Changed/Unchanged
unchanged unchanged
unchanged unchanged
VASA
VASORUM
Intima/Media/adventitia
adventitia
adventitia
adventitia
intima + adventitia
Outer/inner Border adventitia
outer inner/outer
inner inner
N=normal,
AN= between N and A, A=concentrically enlarged,
E=eccentrically changed coronary artery
Morphometrically
intimal-, medial and adventitial-width was
assessed and to make different sized vessels
comparable the relative figures were used:
thickness intima/r-lumen, thickness
media/r-lumen and thickness adventitia/r-lumen.
This resulted in the following mean figures of
Th-int, Th-med and Th-adv in micrometers:
Coronary
artery N
AN A
Th-int
23 136
199
Th-med 77
136 141
Th-adv 120
185 211
In
this way coronary arteries N, AN and A can be
identified in numbers.
The
aim of the follow up study was to investigate
coronary artery remodelling not only in vessels
N, AN and A but also E. Arteries eccentrically
enlarged can be divided into vessels containing
a part N opposite of the plaque (EN), a part A
opposite the plaque (EA) or the plaque is
circumferentially (EE). Serial radial
measurements were performed and Th-int/r-IEL was
used in stead of Th-int/r-lumen
as a measure of the arterial pathology.
The following figure shows the correlation
between the histological- and measured
classification.
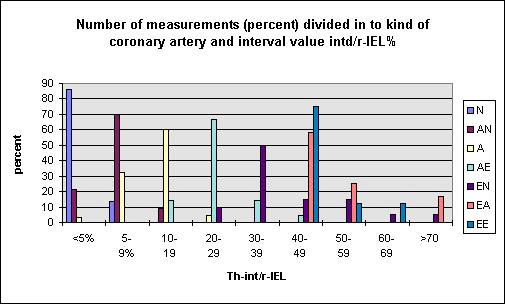
It
appears the correlation is rather good. The data
of Th-int/r-IEL resembles the atherosclerotic
process: normal arteries <5, concentrically
changed arteries 5-19,9. 20-49,9 the arteries
change from concentrically enlarged into
eccentrically and the last phase >50
destruction of the arterial wall and closure of
the lumen (TH-int/r-IEL 100). In this way 4
stages have been defined. The next figure shows
the mean values of r-lumen, Th-int, Th-med and
Th-adv for each stage.
?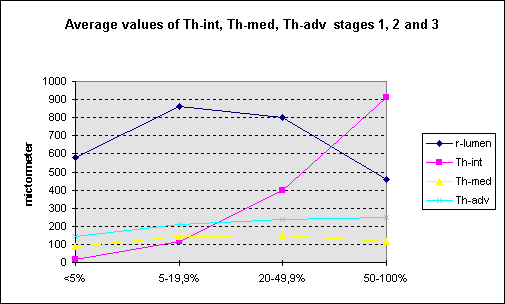
This
figure shows the increase of intimal width
(Th-int/r-IEL 5-19,9) accompanied by growth of
the media and adventitia and also r-lumen
(strong positive remodelling). Or is the
increasing media-adventitia width accompanied by
intimal growth? In any case the first stage of
atherosclerosis (pre-atherosclerosis?) concerns
the entire coronary arterial wall which finding
I can not relate to “atherosclerosis starts in
the intima”.
The
focal nature of the atherosclerotic process
becomes clear when the segments Th-int/r-IEL
<5 – 19,9 from
coronary arteries E are compared to the vessels.
The next table show the result.

The
vessels and segments seem to be similar and so
atherosclerosis affect only a part of the vessel
wall which proves the focal nature not only
lengthwise but even circumferential.
Top
Eddie
Vos
I
just finished first reading of Paul's entire
dissertation [thank you for the copy], a
handsome histology text indeed. My initial take
home impression was that intima enlargement was
always found, and I thought early, but that
outer changes happen also.
I explained this as possibly caused by a decline
of the extra-cellularmatrix, i.e. the elastins,
collagens and associated cartilage
[proteo-glycans], but the dissertation did not
deal with the chemistry of these structural
components. If this comes first [and as per
Kilmer] through the myriad ways by which
homocysteine attacks these structural layers,
such would explain I think the observations of
Paul. Since there is MORE structure in the
intima than in the media [cells and elastin
structure] and adventitia [structure, cells,
fat, vasa vasorum et al, it makes sense that the
health of intima structure is vital and an early
factor.
Things crystallize in one's mind when realizing
that both collagen and elastin are kept together
by lysine based linkages, and that homocysteine
messes with the lysine AND deactivates the
enzyme needed for the linking [lysyl oxidase],
and that nothing much happens in linking without
sufficient vitamin C and copper.
I was not clear if the vasa vasorum [the vessels
of the vessels .. feeding cells, not 'dead'
structure like collagen] that Paul looked at was
mainly the 20-50 micron diameter type that does
not 'invade' the media leave alone the intima
[only after elastic barrier fibers or net- and
sheet-like elastins are destroyed] and if the
vasa vasorum looked at included the 5 micron
diameter network of capillaries [marginally
smaller than a flat red cell but >150x larger
than LDL].
Paul does not say what is in an intima at age 12
but it seems that more cells appear after age
20, his 2 earliest samples of autopsy hearts.
Others have suggested that the intima is
initially essentially a cell free ayer -only
structure, and thus not in need of a permanent
blood supply, and not needing an inflammatory
defense mechanism.
I would imagine that the dozens of concentric
muscle cell layers of the media of larger
arteries -separated by a layered network of
elastins- does need some capillary blood supply
at some early stage. Paul finds that vasa
vasorum infiltrates towards the inside only when
structural layers -internal elastic membrane for
example- are destroyed.
May I refer you to the first paragraph of
http://www.health-heart.org/comments.htm#11
and solicit comments and to the last image on my
home page http://www.health-heart.org
where I attempt to put things together as to
cause. What should I change?
Top
Paul
de Groot
The vascularity of the media has been debated
fiercely in early days. The arterial media of
several animal species contain vessels. However,
the anatomy of the media is also different:
laminated. Vessels are localized between the
lamina. The human coronary arterial media is
avascular that is to say I have never seen any
vessels using light microscopy. So the structure
of the "organ" vesselwall is peculiar
consisting of two avascular layers (intima,
media) and one vascular layer (adventitia). With
that the nutrition of the intima and media
depend entirely on diffusion. Although peculiar
it is not uncommon for example the eye's cornea
and cartilage. It is said the intima plus 1/3
media are dependent on diffusion from the lumen
and the other part from the vasa vasorum in the
adventitia. The nutritian of Eddies poor myocyte
in the middle of the media is in fact subject to
diffusion distance and diffusion gradient, a
process easily disturbed. To keep vessel wall
homeostasis with an increase of vessel wall
width vasa vasorum increase in number and size
and grow towards the lamina elastica externa
minimalising diffusion distance. This repair
mechanism is of use for as long as EEL and media
are intact (Th-int/r-IEL 5-49,9). Further
increase
of intimal thickness and with that Th-in/r-IEL
is accompanied by media destruction and vasa
vasorum growth into the plaque. By now I will
not comment on the cause of this process
(homocysteine?). At
first I want to sow doubt about the primary role
played by the intima in the process of
atherosclerosis.
Top
Uffe
Ravnskov
As far as I have
understood from your dissertation, your
mathematical data gave no support to the idea
that the primary event in atherosclerosis is
damage to the adventitial vessels. But then, why
do you ask: Does it really start in the
intima??? Your finding that all vessel walls
increase, not only the intima, does not
contradict that plaque formation starts from the
inside, because in your calculations you have
only used data from normal vessels and from
vessels with concentric stenosis and excluded
those from vessels with eccentric ones. As you
suggest yourself, eccentric and concentric
stenoses most probably have a different
pathology. Furthermore, it has to be proven that
concentric stenosis has any clinical importance.
Isn´t it the eccentric stenosis that matters,
the plaque, or better the vulnerable plaque, or
the
acute thrombosis? And whatever term you use,
these are situated just beneath the endothelium,
a strong support to the view that
atherosclerosis starts from the inside, at the
endothelial surface.
Top
Paul
de Groot
The vascularity of the media has been
debated fiercely in early days. The arterial
media of several animal species contain vessels.
However, the anatomy of the media is also
different: laminated. Vessels are localized
between the lamina. The human coronary arterial
media is avascular that is to say I have never
seen any vessels using light microscopy. So the
structure of the "organ" vesselwall is
peculiar consisting of two avascular layers
(intima, media) and one vascular layer
(adventitia). With that the nutrition of the
intima and media depend entirely on diffusion.
Although peculiar it is not uncommon for example
the eye's cornea and cartilage. It is said the
intima plus 1/3 media are dependent on diffusion
from the lumen and the other part from the vasa
vasorum in the adventitia. The nutritian of
Eddies poor myocyte in the middle of the media
is in fact subject to diffusion distance and
diffusion gradient, a process easily disturbed.
To keep vessel wall homeostasis with an increase
of vessel wall width vasa vasorum increase in
number and size and grow towards the lamina
elastica externa minimalising diffusion
distance. This repair mechanism is of use for as
long as EEL and media are intact (Th-int/r-IEL
5-49,9).Further increase of intimal thickness
and with that Th-in/r-IEL is accompanied by
media destruction and vasa vasorum growth into
the plaque. By now I will not comment on the
cause of this process (homocysteine?). At first
I want to sow doubt about the primary role
played by the intima in the process of
atherosclerosis.
Eddie
Vos
So, intima and 1/3rd of the media is
nutrient and signal supplied by diffusion from
the inside and the balance of the artery wall
from the outside. So, are you saying that IF
there would have been 5 micron diameter
capillaries in the media, you would have seen
them -and that BEFORE the EEL [external elastic
lamina/layer] deteriorates around the media,
there is no infiltration of capillaries into the
media?
Top
Paul
de Groot
I
owe several of you answers to questions posed on
my, perhaps, provocative saying “does
atherosclerosis really start in the intima???”
I tried to sow some doubt on the subject
and now will try to be more specific. Of course
Uffe the fact that all three layers of the
coronary vessel wall thicken at the same time
does not imply the cause is not in the intima.
My supposition is “the start of
atherosclerosis is in the adventitia”.
Now you all frown and accuse me of
fantasy. Here me out! In literature much
evidence is available on the subject. Nakata (1)
showed experimentally (1967) obstruction of vasa
vasorum cause intimal lesion similar to early
atherosclerotic lesions. Gutterman (2) induced
intimal and medial hyperplasia by dissecting the
adventitia from arteries while in the rabbit (3)
removal of the adventitia intitiates intimal
proliferation and regression of the lesions on
regrowth of the adventitia. The same phenomenon
is seen with positioning a hollow silastic
inflatable collar around the artery (4). At the
other hand an external collar inhibited balloon
induced intimal hyperplasia (5) while in a pig
model external stenting reduced medial and
intimal thickening and growth factor expression
(6). All right you say all these experimental
findings have nothing to do with the
“normal” process of atherosclerosis and
manipulating endothelium can also induce
atherosclerotic like lesions. How about
adventitial fibroblasts migrating to the intima
and playing a role with neo-intima formation
(7). I will not tire you with a full survey of
literature much more can be found.
In
my opinion arterial wall hypoxia, disturbance of
vessel wall homeostasis, could explain the
findings. Hypoxia inducible factor can cause
arterial wall hyperplasia. So if I am right the
arteriosclerotic process to start in the
adventitia the first question you ask is what
changes the adventitia? My answer would be
inflammation. If you can digest the foregoing I
can state my case on inflammation an other time.
The
normal coronary artery can change in different
ways: concentrically enlarged and later on
eccentric grow or directly from normal into
eccentric plaque. I did measure eccentric
coronary arteries Uffe as was shown in one of my
attachments. I did not emphasize then that
coronary arterial remodelling is Th-int/r-IEL
dependent and in this way positive- and
negative- remodelling can take place in the same
artery. This could provoke plaque instability.
With these plaques inflammatory infiltrates are
found at the shoulders of the plaque but
situated in the adventitia. As a matter of
course I do not negate the role played by the
endothelium but I see that as secondary to the
adventitial process. Perhaps that also is the
answer to Leslie: adventitia induced hypoxia as
a result of inflammation give rise to HIF
(hypoxia induced factor) after which the entire
process starts.
- Nakata
Y e.a. Vascular lesions due to obstruction
of the vasa vasorum. Nature 1966;212,
no.5097
- Gutternan
DD e.a. Adventitia-dependent influences on
vascular function. Am J Physiol Haert Circ
Physiol 1999;277(4):1265-1272
- Barker
SG e.a. The adventitia and atherogenesis:
removal initiates intimal proliferation in
the rabbit which regresses on generation of
nea adventitia. Atherosclerosis
1994;105(2):131-144
- Loo
van der B e.a. The adventitia, endothelium
and atherosclerosis. Int J Microcirc Clin
Exp 1997;17(5):280-288
- Fogelstrand
P e.a. External collar inhibits
balloon-induced intimal hyperplasia in
rabbits. J Vasc Res 2002;39(4)
- Dheeraf
M e.a. External stenting reduces long-term
medial and nea intimal thickening and
platelet derived growth factor expresson in
a pig model os arteriovenous bypass
grafting. Nature Medicine 1998;4(2)
- Saverio
S e.a. Contribution of adventitial
fibroblasts to neaintima formation and
vascular remodelling. Circ Res 2004;89:1111
Top
Bogdan
Sikorski
Dear Paul - Even though I got somewhat lost in
your initial paper (too many abbreviations and
tables for my liking), I am firmly with you on
that hypothesis, and as I have indicated to the
group on occasion, I have strong reservations
about the source of the cholesterol in the
arterial wall being blood from its lumen - I
propose it is made right there - in situ. As has
been shown, BBB is virtually impervious to
cholesterol (brain has to make it on the spot),
and BBB closely resembles endothelial lining, if
I correctly remember.
As I have mentioned recently, I have seen
presentation(s) on external arterial cuffs
causing atherosclerotic-like changes in rabbits,
including accumulation of cholesterol and
various cell infiltrates, all because adventitia
and/or vasa vasorum were disrupted/cut.
Again, as shown by Stout in 70s, glucose and
acetate showed up in aortal wall as cholesterol.
Glucose is an excellent oxygen donor, and
cholesterol is a "healer" in the
oxidative stress, as in inflammation. All fits
nicely!
I my previous life, I was vivisection
pharmacologist and cannulation and isolation of
small arteries (carotid; renal) and aorta was a
breeze! Oh, I almost feel like doing it again to
those poor ratties and piggies.
Have you tried in your experiments on animals,
assuming you have done some, to find out if when
a cuff is applied to the artery, a statin given
IV makes a difference in the cross section
picture and cholesterol accumulation of the
artierial wall?
Top
Paul
de Groot
Dear Bogdan, that makes two believers. I
agree fully the puzzle pieces fit nicely if the
atherosclerosis story is told starring the
adventitia. Unfortunatally I did no animal
experiments, in a hospital animals are not
desirable. Formerly tissue samples could be
obtaind easily from obductions and so all my
investigations concern humans. Perhaps a next
time I will try to describe my hypothesis as to
the cause(s) of adventitial changes.
Bogdan
Sikorski
Top
Björn
Hammarskjöld
Why
use cuffs to cause atherosclerotic-like changes
in rabbits? Isn't it enough just to have a high
level of glucose in the vessel?
See
Josephine M. Forbes et al in Diabetes
51:3274-3282, 2002
and other papers like
S. Kooptiwut et al.High glucose-induced
impairment in insulin secretion is associated
with reduction in islet glucokinase in a mouse
model of susceptibility to islet dysfunction. J.
Mol. Endocrinol., August 1, 2005; 35(1): 39 - 48[Abstract]
[Full
Text] [PDF].
J. M. Forbes et al. Advanced Glycation End
Product Interventions Reduce
Diabetes-Accelerated Atherosclerosis. Diabetes,
July 1, 2004; 53(7): 1813 - 1823.
Glucose binds nonenzymatically to proteins and
destroys the thee dimensional structure as well
as changing water solubility.This can cause cell
damage which in turn cause vessel wall damage
and atherosclerosis. THINCs about it!
Top
Uffe
Ravnskov
I
doubt
that anyone in our group believe that plaque
cholesterol comes from blood cholesterol sieving
or transported through the arterial wall. My
problem with the idea that the primary damage
happens in the adventitia is purely based on
common sense. Isn´t the main reasons why
arteries are atherosclerotic but not veins the
intraluminal pressure? And I assume that the
pressure in the adventitial capillaries is only
a little higher than the venous pressure. What
is wrong with Duguid´s hypothesis (see my
previous letter)? To avoid misunderstandings –
I do not think that the high pressure is the
main cause or that high pressure by itself
causes atherosclerosis; rather that it
predisposes to damage from whatever cause.
Top
Paul
Rosch
Uffe: I agree. Sustained hypertension
causes stroke but is an associated "risk
marker" rather than a causative "risk
factor" for coronary atherosclerosis and
the same is true for cigarette consumption,
which can cause cancer of the lung and
emphysema. The MRFIT study clearly showed that
lowering elevated blood pressure, cholesterol
and reducing smoking alone or in combination did
not lower the rate of heart attacks. On the
other hand, heart attacks were higher in men
with certain Type A traits and the WCGS clearly
showed that Type A behavior as assessed by the
structured personal interview was as significant
a "risk factor" for CHD as
hypertension, cholesterol and smoking and was
also completely independent of these. What is
important about this observation is that
although hypertension is not a hallmark of Type
A, such individuals do show hyperactive and
exaggerated blood pressure responses to
stressors and it is these repeated surges that
probably damage the intima and predispose to the
development of plaque. As Jim Lynch and I have
shown, everybody's blood pressure spikes as soon
as we start to speak and the magnitude of this
is affected by speed and volume of speech, the
perceived relative social status of the
audience, the content of the conversation,
presence of a pet and other factors. Although
these elevations can be alarming at times,
patients have no perception of this and are not
aware whether their blood pressure is high,
normal or low. The higher the resting blood
pressure, the greater the rise when you start to
talk. No antihypertensive medications are
capable of blunting these surges and beta
blockers actually accentuate them. Conversely,
blood pressures fall below basal levels when one
is listening to someone else or is silent and
attending to something in the environment, such
as watching tropical fish in a tank. Note also
that the customary rise in blood pressure with
age is not seen in secluded orders of nuns who
rarely speak and occupy themselves mostly by
tending to their plants or crops.
Type A's are poor listeners because they tend to
think about what they are going to say next and
when to interrupt others in order to emphasize
their own points. One of the most defining Type
A traits are vocal stylistics and speech
patterns that include rapid, forceful and
"plosive" speech that results from
inhaling large amounts of air and expelling it
while talking to provide further emphasis. These
"plosive" and other speech
characteristics seen in Type A's not only lead
to proportionately greater increases in blood
pressure while talking but blood pressure also
fails to fall back to basal levels when they
stop because instead of listening they are
thinking of what they are going to say next.
Thus, they are caught in an upward spiral of
increasing blood pressure surges the longer they
continue to talk or try to communicate with
others. Most physicians are unaware of this
since silence is built into the auscultatory
measurement of blood pressure with a stethoscope
but is readily demonstrated with the Dinamap we
use or other automated computerized devices.
Similarly, ambulatory monitoring studies confirm
that the highest blood pressures are seen while
talking to someone and especially during phone
conversations discussing something that is
controversial or stressful.
Deaf mutes show the typical blood pressure surge
as soon as they start to communicate with
someone by sign language but not when they move
their hands vigorously in a meaningless fashion.
The only exception is seen in schizophrenics,
who also tend to be hypotensive. The explanation
for this is too complex to discuss here but
there is a detailed discussion in The
Language of the Heart that also reviews our
research results noted above.
Jim and I are quite confident that the link
between Type A and coronary heart disease will
prove to be these repetitive spikes in blood
pressure that damage the inner surface or
adventitia of coronary vessels that result from
Type A vocal stylistics and poor listening
habits. These and some of our other research
findings have led to the development of a very
successful non pharmacologic treatment for
hypertension by teaching patients to get in
touch with their feelings and how to reduce
blood pressure surges while talking.
Top
Leib
Krut
The idea that cholesterol from plasma
could contribute to cholesterol in the
atherosclerotic plaque via the adventitial
capillaries is not tenable. It has long been
known that the inner half to 2/3 of the media is
devoid of capillaries ( and probably lymphatics)
and at least one of our contributors has
recently pointed this out. This is unlike the
situation in veins, even including the pulmonary
artery, which do indeed contain capillaries, and
probably lymphatics, across the whole of the
media. When there is pulmonary hypertension
(among other things probably causing compression
of the low pressure capillaries and lymphatics
in the inner wall) the pulmonary artery becomes
susceptible to atherosclerotic lesions.
One may well ask
how the inner arterial wall derives its
nutrients. The consensus is, as has again
recently been pointed out by ay least one
member, that it is by the diffusion of plasma
constituents outwards from the lumenal
surface, driven by the high intralumenal
pressure. A nutrient that might be of special
interest for us is LDL. How do the cells in
the inner media of arteries, and every other
tissue for that matter, obtain access to this
essential nutrient. LDL clearly cannot
traverse normal endothelium to reach the
subendothelial space, and beyond that in the
case of the artery, in an intact state. We all
know that when LDL enters a cell, including
the endothelial cell, whether lining an artery
or a capillary, it is extensively degraded.
This thought does not seem to have been
emphasized, but the inference is clear. For
intact LDL from plasma to reach any cell in
any tissue it must by-pass the endothelial
cell. We have in fact long known that this
must happen because LDL (and HDL) is found in
lymph draining peripheral tissues and the LDL
flow is increased when capillaries are
damaged.
Again, we all know that cholesterol
homeostasis in the hepatocyte is critically
determined by the LDL that is delivered to it
from plasma. The hepatocyte obtains access to
intact LDL from plasma by virtue of fenestrae
in the endothelium lining the hepatic
sinusoids. These fenestrae are large enough to
allow transfer even of chylomocrons from their
lumen to the space of Disse. This unique
anatomical arrangement, which allows intact
LDL direct contact with the hepatocyte via the
space of Disse, could serve to emphasize that
LDL must by-pass endothelium if it is to reach
any cell intact, with the obvious exception of
the endothelial cell. And every cell must have
access to LDL from plasma if it is to meet its
own cholesterol needs, unless what Brown and
Goldstein have taught us is nonsense.
I might add that there has been lots of
evidence over the years that the transfer of
constituents from plasma into the normal
artery via its lumenal surface is increased at
sites that are prone to develop
atherosclerotic lesions.
Granting that LDL from plasma gains access to
the sub-endothelial space of arteries via its
lumenal surface, the issue as I see it is
this. If cholesterol is indeed atherogenic,
what is it that converts this normal plasma
constituent into a pathogenic moiety in the
artery?
I would add that the susceptibility of the
pulmonary artery to atherosclerosis when there
is pulmonary hypertension might depend both on
an increased transfer of plasma constituents
into its subendothelial space and to the
obliteration of lymphatics and capillaries
normally present in this region, which would
eliminate the clearance pathway for plasma
constituents not taken up by cells
Top
Paul
de Groot
Uffe, of course it is unbelievable
cholesterol is transported from the adventitia
to the intima and is not what I mean. What I
really believe is (primary) adventitial
inflammation resulting in (secundary)
permeability of the endothelium after which
(perhaps) ox-LDL enters the intima etcetc. Soon
I will try to compose my hypothesis.
Top
Eddie
Vos
I hate to be a 'stick in the intima" and
while swellings anywhere can obstruct and cause
pain, angina is just a warning and does ot kill.
It is important to know where decline and
blockages start but more important WHY, and it
is only when the STRUCTURE of the intima breaks
open, an engineering problem, that one initiates
a heart attack. From page 2 of PWF Wilson's
'Atlas of Atherosclerosis' re the intima of the
'normal' artery: "1.) the endothelium
resting on a thin basal lamina; 2.) the
subendothelial space containing thin elastic and
collagenous fibers and proteoglycans and; 3.
longitudinally oriented smooth muscle cells in
large muscular arteries." [the latter would
be the 'musco-elastic' layer of the intima as
per another 'atlas']. My point and I believe
Kilmer's would be that as long as you don't
damage the 3 components of item 2 in the
'subendothelial space' with homocysteine
[thiolactone], your collagen and elastin should
never fragment and weaken and cells and repair
mechanisms have no reason to move in and try to
remedy. I think the issue is: what is the
precise composition of the intima of a
micronutrient replete (representing homocysteine
<~6 µmol/L) twelve [12] year old. The trick
then is to maintain that precise structural
engineering masterpiece through out life and to
keep what is meant to be acellular just so. What
happens in the adventitia would not be able to
cause structural failure (infarct) if the
structural layers to the inside remain in tact.
That is also true re the collagen fiber and
elastin (layers & fiber) that position the
cells in the media, the structure that is
similarly attacked by the slow protein degrading
homocysteine [thiolactone]. Then, logically,
cells get triggered to move or replicate and
then require more capillaries and (re)generate
the extra cellular matrix upon which they
depend. This is impossible to do well when high
homocysteine prevents the integral lysine
linkages [directly when the molecule below opens
at X or by destroying lysyl oxidase -and m.m.
for proline in collagen]. My final point would
be that homocysteine but nothing else explains
everything historic, i.e the rise-and fall of
CHD with heart attacks first described in dull
detail in JAMA 1912, i.e. after the first
several decades of micro-nutrient removal to
generate the long-shelf life products we have to
day, and the very year the first cookbook for
hydrogenated fat was published [Crisco by
Procter and Gamble].
Top
Bogdan
Sikorski
OK Eddie - Again,
nicely put! and convincing.
Just a simple question - has anyone stuck a
radioligand on homocysteine and found it where
you and Kilmer say it should go when present in
a relative abundance (>6 µmol/L )?
Top
Paul
de Groot
Uffe,
Duguid
published a lot of micrographs of microthrombi
covered by endothelium but AFTER a heart attack.
That is essential. A heart attack (myocardial
infarction) is a result of blockage of the
coronary artery by plaque bleeding c.q. plaque
rupture. In both cases thrombi are formed on the
bulging mass in the coronary lumen: flow
stagnation. The thrombi are incorporated in the
plaque and the endothelial lining is restored.
The thrombi fibrose and often recanalization
takes place. Sometimes such a plaque is
laminated as a proove of more than one thrombus.
This picture is definitely different from the
"normal" atherosclerotic plaques which
show a fibrous cap on a mass of undifferentiated
tissue (pultaceous mass) in which blood vessels
(from the adventitia), spiked spaces seen as
cholesterol crystals??, fibrous and collagen
connective tissue etc. It shows more or less the
composition of scar tissue. This is the reason
atherosclerosis is now described as a chronic
inflammatory disease. So thrombosis and
atherosclerosis are different processes,
however, can go together.
With
that your reasoning thrombosis, fatty streak,
fibrous plaque, complicated plaque is highly
unlikely and
in comflict with facts.
Top
Uffe
Ravnskov
Paul,
the
crucial question is, what causes thrombi and
what causes plaque bleeding? Here is Duguid´s
interpretation of his findings:
“Microthrombi
occur at as early as three years of age and
continue to occur throughout life. By forty
years their repeated incorporation into the
intima results in irregular fibrous thickening
which are too stiff to comply in the normal way
with pulse movements and so cause disruption and
haemorrhage. The extravasated blood
disintegrates leaving fatty deposits, which
accumulate progressively, and when haemorrhages
are frequent and profuse the deposits
predominate whilst the fibrous tissue is
relatively sparse, with the result that the
thickenings are soft and friable. In such
circumstances the surface layers are liable to
be torn away leaving ulcers, which promote gross
mural thrombosis. The thrombi become organised
and form fibrous thickenings which narrow the
arteries and destroy their elasticity.”
As
I see it Duguid´s hypothesis leaves room for
many factors that may cause the formation of
microthrombi, and also for factors that may lead
to incomplete healing and thus predispose to the
production of soft and friable vascular scars
(eg. plaques).
Top
Herbert
Nehrlich
This seems to beg the question of the apparent
absence of these processes in certain
populations. Could the disturbed (reversed)
ratio of n-6 to n-3 in "modern man" be
the key? I am always reminded of Dr. Paul Dudley
White's (White House Physician under Eisenhower)
statement that he could not find patients to try
his new gadget on, it was the EKG machine.
Surely this isn't all just one big fault in the
design of the human being.
Eddie
Vos
Friends, sure, it's a fault of being human and
having supermarkets and grocery stores around
and there seems little evidence that before
about 100 years ago we got such
mirco-coagualtion problems leading to infarct
and decline.
When animals
eating non-processed and non heated foods with
say 4-5x the homocysteine lowering B vitamins
and none of the refined starch revolution foods
don't seem to have heart attacks/atheroma, this
alone is a message.
There are clearly
2 methods by which we damage long-living
structural proteins: glycosylation from excess
carbs [O-bonding glucose to proteins] and
thiolation [sulfur bonds/insertion in proteins].
Both methods are nutrition related. Nobody would
do animal or culture dish experiments with such
clearly malnourished subjects as in current H.
Sapiens. We're overwhelmed with nutritional
confounders.
With Uffe:
clearly the 'coagulopathy' elements [all
micro-nutrients], and via glucose or insulin
PAI-1 for example, is a fascinating subject and
evidently micro-and macro clots play roles -but
are they primary? Also, in the coagulopathy
department, Herbert's n-3 helps while n-6
clearly does the reverse.
Top
Paul
de Groot
Uffe, To day I am a bit philosophical because
we, al together, seem to discuss different
things. My good friend Roel put it this way. If
you look to our globe from very far off you can
see a town. If looking closer you see a town
with clogged streets and a still closer look
reveals cars in the street for example most cars
are Renaulds and Peugeots. My assumption is the
town is somewhere in France. A look at a street
sign says "champs elisee" and so the
town I saw from far of could be Paris although
perhaps there are more towns with a Champ
Elisee. What clogged the street in Paris?
possibly road work, however, in London the
street is also blogged but possibly by non
working street lights. Although we see in both
towns the same thing, street congestion, the
cause is quite different. This came to my mind
reading your e-mail. Atherosclerosis is the name
we use for arterial wall changes showing
excentrically plaques and we assume the process
is the same in all arterial vessels although we
know many a time there is predilection, cerebral
vessels, peripheral arteries, renal arteries,
coronary arteries and last but not least aorta.
So ..."the surface layers are liable to be
torn away leaving ulcers..." is a statement
clearly concerning the aorta; I have never seen
ulcers in a coronary artery. I have studied only
(human) coronary arteries and so I can only
discuss these vessels. With this in mind I still
can not understand why after many years of life
all of a sudden the endothelium become permeable
for say cholesterol, ox-LDL and/or thrombi.
Let's have a look at the so called (coronary)
complicated plaque. The big difference between
the complicated plaque and the fibrous plaque is
its vascularity, the fibrous plaque contain no
vessels this in contrast to the complicated
plaque. The plaque vessels grow from the
adventitial vasa vasorum via a destructed media
into the plaque. The vessels are thinwalled,
form no pattern and are irregular of size and
are prone to disruption according to the many
forces executed. The result is plaque hemorrhage
and subsequently forming of a cholesterol pool
which we later on can see as cholesterol
chrystals. With this process external
cholesterol does not play a role.
My proposition is to abandon
"atherosclerosis" and make up new
names describing the process at hand in the
various sites and with that make discussions
more to the point. Any suggestions?? Perhaps my
argument sound a bit pedantic, if so I am sorry,
Eddie I rode my "stokpaard" again.
Top
Eddie
Vos
Hi Paul, just a note since you mentioned my name
in your response .. how about
athero-arteriosclerosis .. of simply 'artery
tire failure', artery puncture or re the aorta
'ply delamination' since you made that nice
vehicle traffic jam analogy? Let me suggest that
it is slow decline of structure that allows your
vasa vasorum in, but that the decline of
structure comes first ...
Let's realize the importance of the structural
components like elastin and how it degrades and
allows calcium and lipid deposits in that broken
structure [in later type V, VI lesions]. This
attachment of exceptional length [sorry] is a
2005 article from Atherosclerosis that
wonderfully describes the process of decline in
the inner half or so of the artery: great
electron microscope work indeed. This article,
read together with the 2000 J. of Nutr. Carlos
L. Krumdieck article I sent around about 2 weeks
ago, is required reading when one wants to
understand the more fundamental processes at
play. Re Krumdieck: Melchior commented to me: I
finally understand the protein decline theory.
The Yuri V. Bobryshev article uses terms as
'vacuolization', holes or 'cavities' in elastin,
'crumbling' elastin, 'splitting of elastin fiber
fragments' ... guess what, homocysteine
(thiolactone) will do that! Sorry about riding
my my own 'stokpaard' [hobby horse].
Top
Paul
de Groot
Hi Eddie, I agree to the importance of the
structural components of the arterial wall.
However, why degrades elastin it has to have a
cause. We all know that sclerotic changes of the
arterial wall is not per se age dependent and so
is not a fact of life. There ought to be one or
more causes and possibly the causes are not the
same for different arteries. Why would elastic
arteries change in the same way as muscular
arteries from the same agens? and why would a
different "organ" such as the aorta
react in the same way. We just don't understand
the differences and still we think that there is
just one disease called atherosclerosis which is
responsible for the changes of all arteries.
This is inconceivable, we still can only see
clogged streets in a town unknown and so we must
try to acquire the knowledge which town in which
state and what caused the jam there. Still your
attachment is very interesting but the
conclusions are only valid for carotic arteries.
I still try to read and understand your papers
on homocysteine, however, my chemical knowledge
is rusty and from a long time ago but
"nooit te oud om te leren".
Top
Eddie
Vos
Right, we're evidently as rusty [crumbly] as our
arteries but as you said never too old to learn.
Maybe it's more the elastin that comes apart
since it ENTIRELY depends on the mirconutrient
dependent [Cu, vit. C] and Hcy-lactone degraded
X-linking system, while collagen will still
function somewhat in the light of the same
factors. I can see that collagen 'rope' still
pulls well yet in pushing mode a rope may come
undone and it's there that the OH-lysine /
OH-proline keep the strands together systems
gets stressed, at least that is my simplified
way of looking at it. What a great term in that
study: crumbling elastin. Now, let's jump back
to Kilmer's Classic of 1969: 'splitting,
irregularity and focal discontinuity of the
internal ELASTIC membrane' ..
Top
Uffe
Ravnskov
I have a few questions and objections.
To Paul: I am still sceptical to the idea that
the primary event is inflammatory reactions of
the adventitial vessels. If intima and inner
media receive their nutrients and oxygen by
diffusion from the lumen, what role does these
inflammatory processes play for the creation of
endothelial lesions? Couldn´t the experimental
studies, where the authors hurt the adventitia
or its vessels be explained otherwise? Saul and
Gerard presented an interesting hypothesis a few
years ago (Med Hypotheses 1991;36:228-37; ibid.
1999;52:349-51). They considered extra-arterial
pressure as a crucial factor. If it is low, the
artery wall increases its tension or resistance
to prevent its lumen to expand, either by
fibrosis or by deposition of other rigid
material such as cholesterol. They claimed for
instance, that arteries located in bone channels
never become atherosclerotic, neither do the
small coronary branches that go perpendicular
through the myocardial wall because there is no
need for strength here. Their hypothesis means
that at least some part of the arterial changes,
in particular the fibrosis and the growth of the
muscular wall, are normal features to prevent
dilatation of the arteries and unnecessary
pumping of the heart.
Therefore,
if you damage the adventitia the outer muscular
wall may not be able to maintain a sufficient
tension and thus give rise to endothelial
damage.
Still,
as I see it, this mechanism seems possible, but
unlikely, biologically seen. Why should
adventitial vessels become inflamed? Isn´t it
much more likely that the endothelium does,
exposed as it is to much higher pressure
gradients and dynamic forces and therefore
predisposed to become damaged by all the toxic
chemical factors we have mentioned so often?
By
the way, Saul and Gerards hypothesis also
explain why the coronaries are more prone to
atherosclerosis than other arteries because they
are exposed to an extravascular negative
pressure (as is the aorta).
I
have a question to Leib also. In a previous
letter you claimed that LDL “as we all know”
is extensively degraded when it enters a cell.
As far as I have understood, LDL is not able to
pass any cell wall, neither is it degraded when
it comes into contact with one. Please explain.
You
also wrote that every cell must have access to
LDL from plasma if it is to meet its own
cholesterol needs. First, do you mean
LDL-cholesterol? If so, why call it
LDL-cholesterol instead of just cholesterol.
Second, I have learnt from more knowledgeable
colleagues that any cell is able to synthesise
cholesterol. Isn´t that true any longer?
Paul
de Groot
Who told you about inflammation of
adventitial vessels? Not me! I wrote about
inflammation of the ADVENTITIA of coronary
arteries. In fact adventitial infiltrates are
mostly found around vasa vasorum and adventitial
nerves but also often in surrounding fat tissue
and even very often in epicardial tissue. My
investigation showed adventitial infiltrates in
10% of normal coronary arteries, 48% with
concentrically enlarged vessels, 55% with
"fibrous plaques" and 86% with
complicated plaques which corresponds with resp.
thickness intima divided by radius
lumen+thickness intima (r-IEL) of <5%,
5-19,9%, 20-49,9% and >50%. This in contrast
to plaque c.q. intima infiltrates:
th-int/r-IEL<5%: 0%, 5-19,9%: 21%, 20-49,9%:
26% and th-int/r-IEL>50%: 91%. So even with
complicated plaques (th-int/r-IEL>50) 9% of
plaques are without infiltrate. The correlation
between adventitia infiltrates and plaque
infiltrates is poor. These figures are valid
only for epicardial coronary arteries. As you
mentioned the arteriae perforantes which run
perpendicular to the myocardium are seldom found
pathological, however, these vessels are small,
radius about 200micrometer and so are near the
size of arterioles. Adventitial thickness of
arterioles in proportion to radius lumen is
considerably larger than with epicardial
coronary arteries and so are not comparable.
As I wrote before
inflammation of the coronary vessel wall, at
first the adventitia, is in my opinion the start
of the arteriosclerotic process. Perhaps I was
not clear on this not an acute infection by
bacterial or viral antigens but a delayed type
"allergic" reaction after an airway
infection. A certain similarity to Reumatoid
arthritis and LE in which coronary artery
disease occurs far more often. Every new
viral/bacterial infection give rise to new
arterial damage. This accounts for angina to
become unstable and after a while stable again
with or without medication.
One last thing.
Saul and Gerards hypothesize why coronary
arteries are more prone to atherosclerosis than
other arteries. Unfortunately I have no recent
figures on this but I really doubt coronary
arteries are more prone to atherosclerosis. Only
with FH this is true. Arteriosclerosis can occur
without hypercholesterolemia and damages
arteries sometimes only cerebral, sometimes
renal or carotic arteries, sometimes peripheral
arteries and sometimes coronary arteries and
sometimes combinations. The question is,
however, is the process for all these arteries
the same??
|